Hable con ELA
"Para todos los que han sentido esta enfermedad atravesar sus vidas, Stephen Hawking no solo era un extraordinario científico y divulgador, era el rostro insistente de una sobrevida imposible, el recordatorio de este monstruo incierto, esta enfermedad que existe y acecha".
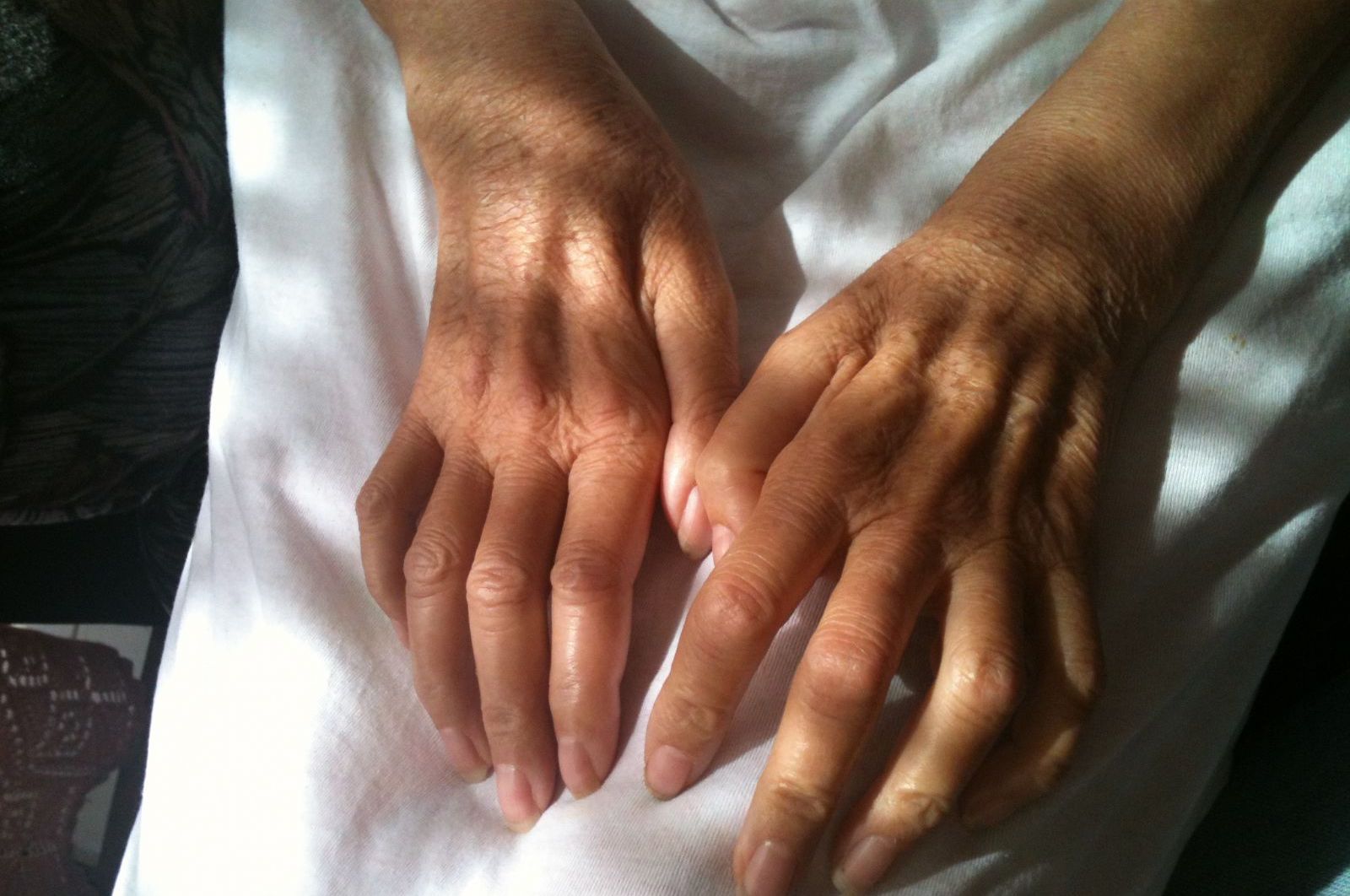
Óscar Marcelo Lazo es Neurobiólogo y Doctor en Fisiología. Investigador en el UCL Institute of Neurology. @omlazo
Una enfermedad impronunciable.
Empiezo a escribir esto cuando acaba de morirse Stephen Hawking, pero me tomo unos días para procesarlo. Hawking no solo ha sido un físico porfiado y absolutamente brillante, sino también uno de los más famosos pacientes de una enfermedad neurodegenerativa llamada Esclerosis Lateral Amiotrófica (o ELA) de la que también sería bueno hablar. La vida de Hawking no fue solo agujeros negros, ideas arriesgadas sobre el tiempo y el inicio del universo, no fue solo logros académicos y reconocimiento. En la voz sintética del computador de Hawking miles de historias de otros afectados por esta enfermedad encontraron un eco humano, una manera de romper el silencio y existir para la opinión pública, visibilizar sus necesidades y acaso convocar a quienes pudieran encontrarle alivio. Y de esa misma manera la noticia de su muerte reactiva la herida de la pérdida para las familias de los caídos y le hiela la espalda a los recién diagnosticados. Porque para todos los que han sentido esta enfermedad atravesar sus vidas, Stephen Hawking no solo era un extraordinario científico y divulgador, era el rostro insistente de una sobrevida imposible, el recordatorio de este monstruo incierto, esta enfermedad que existe y acecha.
Este texto es un esfuerzo por tocar tierra. Despejar un poco la neblina de las palabras incomprensibles que escuchamos decir al neurólogo, hacer una pausa en el terror, la negación y la incertidumbre, poner en orden los resultados de todos esos exámenes que viste o que te has venido haciendo recientemente. Mirar atrás la historia de los pacientes de ELA que hemos conocido o mirar adelante el futuro de los que recién empiezan a quedarse sin fuerzas. Este texto es para mirar al monstruo a la cara y preguntarle quién es.

Ya su nombre, Esclerosis Lateral Amiotrófica, es casi impronunciable y pocos saben exactamente qué demonios significan esas tres palabras que usó el Dr. Jean-Martin Charcot para describirla en 1874. En una época en que ni siquiera se conocía bien la anatomía de las conexiones cerebro-espinales, Charcot estudió diversas enfermedades del movimiento y en algunos pacientes pudo distinguir que ciertos tractos laterales de la médula espinal sufrían un daño que dejaba cicatrices y endurecimiento (esclerosis lateral), bloqueando el movimiento de los músculos y provocando su disminución (amiotrofía).
A mediados de 1939, todo Estados Unidos escuchó mil veces repetidas esas tres palabras, cuando uno de los más extraordinarios beisbolistas e ídolos deportivos, Lou Gehrig, fue diagnosticado con ELA en la Clínica Mayo el día que cumplió 36 años. Después de 17 temporadas en el béisbol profesional, había tenido un sospechoso y brutal declive físico en el último año, que lo hizo mandarse él mismo a la banca de los Yankees. Murió en Junio de 1941. En Estados Unidos la ELA aún es conocida como “Lou Gehrig’s disease”.
Argentina vio quedarse quieto y morir de ELA al extraordinario dibujante y humorista Roberto Fontanarrosa. Tenía unos 57 años cuando fue diagnosticado el año 2003, después de comenzar con síntomas de debilidad en sus piernas. En enero del 2007 dejó de dibujar y en junio de ese mismo año murió de una falla respiratoria. Uruguay vio morir de ELA a Raúl Sendic, fundador de los Tupamaros. En Chile, el Cardenal Carlos Oviedo y el músico Peter Rock, sobrevivieron menos de 2 años después de ser diagnosticados.
Más de 250 personas serán diagnosticadas en Chile durante el 2018. Para mediados del 2020, la mitad de ellos habrá muerto y los demás necesitarán cuidados las 24 horas del día. Los costos para las familias y el sistema de salud son enormes, mayores que frente a cualquier otra enfermedad neurodegenerativa, y solo seguirán incrementándose. Las proyecciones indican que, como consecuencia del envejecimiento de la población, para el 2040 habrá en el mundo 400.000 personas con ELA.

La búsqueda científica.
Decenas de grupos de investigadores básicos y clínicos trabajamos en todo el mundo para tratar de entender diferentes aspectos de esta enfermedad. Pero no ha sido fácil. Desde el principio ha sido evidente la enorme variabilidad que la enfermedad muestra. Mientras Lou Gehrig vivió un par de años de rápido deterioro, Stephen Hawking progresó lentamente y sobrevivió más de 50 años después del diagnóstico.
Solamente en el 10% de los casos hay una clara herencia familiar que explica la aparición de la enfermedad y todo el resto es denominado esporádico y sus causas mayoritariamente desconocidas. Sin embargo, ese 10% de ELA familiar hace evidente que en algunos casos basta con una cierta mutación genética para causar la enfermedad. El primero de esos genes fue descubierto por el grupo del Dr. Robert H. Brown Jr. del Massachusetts General Hospital en 1993, y resultó ser el que codifica una proteína crucial para el manejo del estrés oxidativo en la célula, la Cu/Zn superóxido dismutasa (SOD1). Inicialmente se pensó que la oxidación jugaba un rol central y a fines de los años 90 no era raro que a los pacientes se les suministraran suplementos antioxidantes. Pero pronto se descubrió que ese no podía ser el problema, puesto que entre las diversas mutaciones del gen SOD1 que causaban ELA, no todas afectaban de la misma manera su función enzimática.
En todo caso, el descubrimiento de las mutaciones en el gen SOD1 fue un tremendo paso adelante. Ese primer gen descubierto permitió entre otras cosas reproducir la patología en animales de laboratorio y así estudiar los mecanismos de degeneración y potenciales tratamientos de manera controlada. Hoy sabemos que las mutaciones en SOD1 están presentes en un 20% de los pacientes de ELA familiar y explican apenas un 2% de la forma esporádica. Otros muchos y muy diferentes genes causantes de la enfermedad han sido descritos desde entonces, siendo el gen C9orf72 donde se concentran el mayor número de mutaciones —en particular, repeticiones de la secuencia GGGCCC, algo parecido a lo que pasa en la enfermedad de Huntington. Las mutaciones en el gen C9orf72 explican hasta el 40% de los casos familiares y el 7% de los esporádicos, y además son una de las causas más importantes de otra enfermedad neurológica: la demencia frontotemporal. La relación entre ambas patologías es una de las pistas más interesantes que algunos investigadores están siguiendo, preguntándose por cuáles son los mecanismos en común.
A medida que más sabemos, mayor es nuestra conciencia de que se trata de una enfermedad plural y heterogénea. Además de SOD1 y C9orf72, se han identificado mutaciones en otros 15 genes capaces de causar ELA, entre ellos FUS, TDP-43, OPTN, ANG, DCTN1, SQSTM1 y TBK1. Las proteínas codificadas en esos genes tienen diversas funciones: unas participan en el metabolismo del ARN, otras en el transporte de materiales y señales adentro de las neuronas, otras en la degradación de proteínas mal plegadas o que están formando agregados. En ocasiones parece que lo que mata a las motoneuronas es algo que ocurre dentro de ellas —por ejemplo, una falla en la eliminación de agregados de proteínas o defectos en el transporte. Pero otras veces parece que el daño depende de las células que acompañan a las neuronas en el tejido nervioso, llamadas células gliales. Por ejemplo la sobreactivación de unas células de defensa llamadas microglías, una menor eficiencia energética de los oligodendrocitos que forman la vaina de mielina o un envenenamiento de los astrocitos.
En todos esos frentes se está librando la batalla científica por entender la enfermedad y curarla. Todos los días aparecen en promedio unos 4 nuevos artículos científicos sobre ELA en las más importantes revistas del mundo y en lo que va del 2018, ya se han publicado 596 trabajos. Pero no cabe duda de que todavía estamos lejos y necesitamos más esfuerzos, más recursos, más talentos. Investigadores clínicos y básicos comprometidos y con las mejores herramientas para responder a los desafíos conceptuales y a las necesidades concretas de los pacientes actuales. En los mejores centros de investigación del mundo hay creciente conciencia de que la comunidad de pacientes, familiares, investigadores y médicos debe trabajar junta y colaborar de forma estable para que podamos avanzar más rápido.
Tú que tienes ELA.
Estamos todos juntos en esto. La mitad de quienes están leyendo esta columna tiene alguien cercano con Esclerosis Lateral Amiotrófica, o lo tendrá. El riesgo de que nos toque alguna vez en la vida es alrededor de 1 en 350 —el riesgo de sufrir Azheimer, por ejemplo, es mayor a 1:20 desde los 65 años.
Tampoco es cuestión de tener miedo. Más bien lo digo para exorcizar demonios.
Un día sin darnos mucho cuenta podríamos andar con las piernas más torpes o con una mano débil. Esa sensación de no tener fuerza que todos hemos sentido alguna vez en medio de un ataque de risa o cuando recién nos despertamos, se quedará un día entero, y al día siguiente, y después de tres o cuatro días sospechas que algo no anda del todo bien y decides ir al médico. Habrá exámenes, muchos exámenes: radiografías, resonancias magnéticas, tests cardiovasculares, exámenes de sangre. Habrá varios médicos. Y pasarán varios meses de incertidumbre mientras la debilidad se consolida y hasta te empiezas a acostumbrar. Ningún examen da pistas de cuál es la enfermedad: no habrá virus ni infecciones, ni deformidades en la columna, ni tumores, ni anticuerpos que sugieran una enfermedad autoinmune. Sin diagnóstico, llegarás a la electromiografía y sus decenas de pinchazos dolorosos que confirmarán que, pese a que todo lo que te han medido aparece normal, la comunicación entre tu médula espinal y los músculos está seriamente disminuida. Hay menos conducción, respuestas más lentas, asimétricas. Hay daño en los tractos motores.
Al neurólogo que vea el resultado de la electromiografía se le confirmará una sospecha largamente incubada. Te recibirá tratando de disimular que está destrozado, porque ya ha visto el lado más duro del futuro. A esa cita tienes que ir acompañado, porque es ahí cuando te lo dirán y es demasiado peso para un par de hombros ya medios cansados. Aunque a esas alturas no será mucho lo que proceses, te dirán que tus neuronas motoras se están muriendo y que es una enfermedad de causa desconocida, progresiva e incurable. Y rumiarás largamente esas palabras: desconocida, progresiva, incurable.
El golpe vendrá primero cuando te des cuenta que la otra pierna o el otro brazo también están sufriendo debilidad. Te asombrará ver que en unos meses la enfermedad ya avanzó y entonces te caerá la teja. No busques en internet. Preguntarás si tus genes pasaron a los hijos, si los remedios que tomas realmente servirán de algo. Empezarás a pasar revista a tu vida y lo más probable es que no haya estado nada mal, considerando que lo más probable es que todo esto esté pasando hacia finales de tus 50 y comienzos de tus 60 años.
Pero si eres más joven, son otros los fantasmas y tendrás que aprender a domarlos. Lou Gerihg fue diagnosticado a los 36, Hawking a los 22. A estas alturas empezarás a preguntarte si vivirás 2 o 20 años más, a ratos inclinándote más bien por lo primero. La depresión es tu enemigo más urgente y tienes que tomártela en serio si no quieres empezar a morirte ahora mismo. La terapia y los antidepresivos son más importantes que el Riluzole que te recetó el neurólogo. Es verdad que ese fármaco es el único que hasta ahora ha mostrado algún efecto positivo consistente —sobre todo en pacientes con la forma bulbar de la enfermedad. Pero el tratamiento costará más de $5.000.000 al año y no lo cubre el AUGE. La evidencia sistemática muestra que después de desangrar a tu familia para cubrir los 15 mil pesos diarios que cuesta el tratamiento tu probabilidad de sobrevida aumenta en más o menos dos meses. ¡Dos meses! Yo te entiendo si mandas a la mierda el “tratamiento”.
No gastes plata en homeopatía ni te pongas en manos de chamanes, brujos o fantasmas. No importa quién lo recomiende. Todos van a estar desesperados por hacer algo.
Después de algunas caídas y de cierta resistencia, te sentarás en la silla de ruedas y te irás adaptando poco a poco a ser asistido para todo. Te pondrás exigente y algo tirano, pero también encontrarás un modo de habitar tu cuerpo, tu casa, tu vida. La mitad de las personas con ELA mantendrá sus habilidades cognitivas intactas y de algún modo se volverán más agudos, dirán lo justo y necesario porque les cuesta cada vez más hablar y eso se irá convirtiendo en una especia de sabiduría de oráculo. Muchos amigos desaparecerán porque no sabrían qué decir y no saben si lograrán estar contigo sin llorar a gritos, pero también estarán esos que disfrutarán del silencio contigo, esos con quienes el silencio no es incómodo y tu enfermedad será espacio de intimidad y confianza. Habrá tiempo para pensar, mucho tiempo para pensar, y eso hará salir quién más profunda y genuinamente eres: los rollos, los talentos, los terrores y las generosidades.
Te dará rabia, sobre todo cuando la gente que no te conoce te infantilice, te hable fuerte, te trate como si fueras imbécil. Y es ahí cuando se vuelve relevante que tengas voz. Una voz propia o ajena, la voz de Fontanarrosa o de Hawking. La voz de los miles de científicos y activistas que se comprometieron con el ice bucket challenge hace unos años.
Por eso, si tú todavía no tienes ELA o no conoces a nadie con esa enfermedad ayuda a hacerla visible y a que más líderes políticos y culturales escuchen sus voces. Dona tu tiempo, tu trabajo o tu vocación. Ayúdanos a descubrir sus secretos y quizás su cura.
Habla de ello, pregunta a tu alrededor. Porque es bueno que esta enfermedad no sea abstracta, sino que tenga un rostro. Para algunos es el rostro de Gerihg, de Sendic, de Hawking, de una amiga, de un vecino. Para mí es el rostro de mi padre.
¿Cómo quedarse inmóvil?












